
Published by Pirouz Daftarian, Ph.D. on
The goal of cancer immune checkpoint therapies is to cure tumor-specific T cells from dysfunction which is caused by elements from tumor deposits. In this blog, we briefly describe the process of T cell exhaustion and how it can be harnessed for immuno-oncology drug discovery and more specifically to screen immune checkpoint drug candidates. First, the current function-based characterization methods for immunomodulatory drug candidates will be summarized. We have selected three methods which mimic prerequisite parameters of early T cell exhaustion (TEX). Next, we briefly describe an improved recall antigen-based potency assay as detailed elsewhere. Lastly, we describe an in vitro reversible early TEX-like model, which may be tailored with cancer-induced T cell suppression agents. Here it should be noted that cancer induced T cell dysfunction is also referred to as TEX, covering a broad spectrum of different molecular T cell pathologies. The term “T cell exhaustion” in this review mainly refers to an early stage of this process. We have devised an in vitro model that recapitulates components of tumor deposits responsible for inducing TEX.
TEX is a phenomenon that describes a T cell differentiation continuum characterized by hierarchical or progressive diminution of effector function and proliferation, elevated and sustained expression of multiple co-inhibitory receptors, altered homeostatic cytokine response, altered expression and usage of transcription factors, epigenetic rewiring, and dysregulated metabolism. These factors distinguish TEX from T effector (Teff) cells, memory T cells (Tmem) as well as quiescent T cells [1, 2]. TEX cells develop in conditions of persistent antigen and become antigen-dependent as has been shown in the LCMV chronic viral infection model [3], as well as in cancer [4]. Persistent antigen exposure can induce exhaustion not only in CD8+ T cells but CD4+ T cells as well [5]. For cancer, in addition to persistent antigen exposure within the tumor microenvironment (TME), tumor-mediated immunosuppressive metabolic byproducts, suppressive cytokines and cellular debris converge to inhibit T cell function [1, 2, 6, 7]. This combined with increased competition for glucose and oxygen serves to further dampen TEX anti-tumor activity [8]. One such immunosuppressive agent is potassium. Excessive extracellular potassium released into the TME from dying tumor cells can reach a concentration that leads to intracellular potassium T cell influx causing them to become dysregulated [9]. This phenomenon leads to decreased T cell uptake and consumption of nutrients, ultimately resulting in reduced effector gene expression such as IFN-g [10]. Another immunosuppressive agent that is the focus of intense investigation is adenosine. Hypoxic conditions lead to increased extracellular adenosine which suppresses anti-tumor T cell activity via signaling through cognate receptor A2AR [11]. TME suppressive mediators such as these are prevalent in the solid tumor microenvironment and contribute to the resistance of many tumors to immune checkpoint blockade (ICB) [12].
Current in vitro function-based potency assays for immune checkpoint drug candidatesMany factors influence the degree of TEX including antigen-rich environments, un-helped T cells, altered metabolism, ions, lipids, the milieu of cytokines, combinations of these as well as additional suppressive factors [1, 2, 6, 7, 13].
The three commonly used potency assays are Recall Antigen Assay (RAA), Mixed Lymphocytes Reaction (MLR) [14, 15], and the staphylococcal enterotoxin B assay (SEB). The core principles of these functional assays for potential immunotherapeutic biologics are i) these assays can be optimized as a TEX-like assay by inducing co-inhibitory molecule expression, ii) screening assay for active antagonist antibodies to immune checkpoint molecules or agonist antibodies to co-stimulatory molecules with read-outs including antigen-specific T cell expansion, IFN-g secretion, surface expression of activation markers, or IL-2 secretion in MLR [16], SEB, and RAA, and iii) modularity in enabling manipulation of culture conditions to include TME metabolites that induce T cell suppression/dysfunction with similar read-outs as in ii. These three assay types have been utilized by academia and industry for functional characterization of immunotherapeutic candidates from discovery phase all the way to later stages of the drug development [17-19]. Such assays are required to be validated for sensitivity and specificity, accuracy, representativeness of the mechanism of action, physiological relevance, reproducibility, and finally predictivity of clinical efficacy [20]. In addition, these assays may be adopted and tailored to share some aspects of T cell dysfunction seen in the tumor deposit. However, since most ICB drug candidates have unique features but lack enough data for predictability of clinical efficacy, companies employ multiple assays to ensure that in vitro potency assays will translate to the clinic. To this end, similar assays have been used in submissions to regulatory agencies as evidence of non-clinical pharmacological in vitro potency assays (example, FDA BLA # 125554).
MHC tetramer guided functional assays for careening immune checkpoint drug candidates
The added value of analysis of antigen specific T cells (e.g. by using a pool of tetramers) is that one can obtain ample information on these cells. Unlike ELISPOT of ELISA as readouts, cells stained by tetramer maybe stained for all co-inhibitory, co-stimulatory molecules, and /or intracellularly stained for Ki67, CD107a, IFN-g, by gating on tetramer positive / antigen stimulated T cells. MHC tetramer staining also allows for bulk or single cell genomic or proteomic analysis when needed.
Recall Antigen Assay
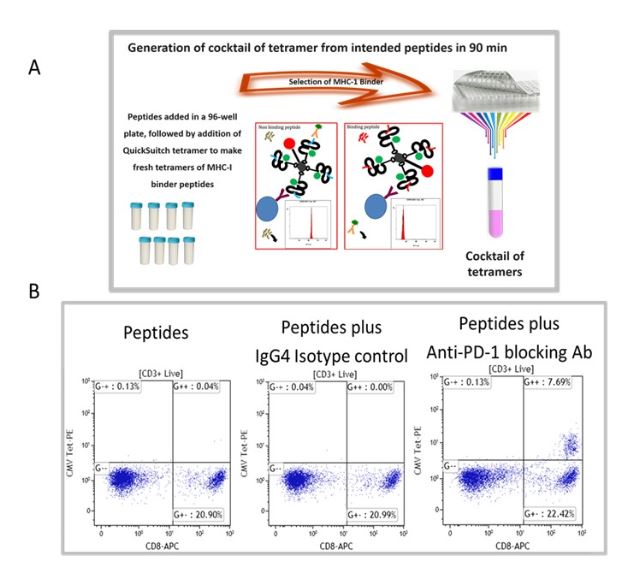
We have selected recall antigen as an assay for screening of ICB drug candidate, for it is more physiologically relevant, as the antigens may be defined and the assay can be conducted in an antigen controlled system, and the MHC tetramers may be used as a readout which allows for obtaining specific information. However, currently there is no standardized protocol and the current status of the assay suffered from poor reproducibility and low signal-to-noise. To this end, we have improved and optimized several parameters of the assay including the stimulation stage, the plate polymer, culture media, interleukins / milieu of cytokines, and finally improving readouts to include cytokines and measurements of expanded recall antigen-specific T cell clonotypes in using a cocktail of MHC tetramers. Figure 1 depicts an improved recall antigen assay for function-based screening of ICB drug candidates. We have selected and characterized donors who have positive recall responses to multiple antigens, for example, those who have low frequency of preexisting CD8+ T cells for CMV, EBV, influenza virus, and WT-1. In doing so, we stimulate with a cocktail of peptides and then detect response with a pool of corresponding tetramers (all may have one fluorochrome). This approach improves the assay’s signal-to-noise ratio and better captures the effect of candidate drugs being screened. To this end, we have used either manufactured tetramers (MBL International, Woburn MA) or as shown in Figure 1A we have used the peptide exchange MHC tetramer platform (MBL International, Woburn MA), which allows the generation of a validated cocktail of MHC tetramers with recall antigen peptides corresponding to multiple recall antigens. The tetramers are pooled after individual production and a desalting step to remove unbound peptides. We have validated this tetramer pool system and the staining pattern was not significantly different from manufactured tetramers for all tested peptides. Figure 1B shows the result of a selected and characterized “PD-1 blockade responder”.
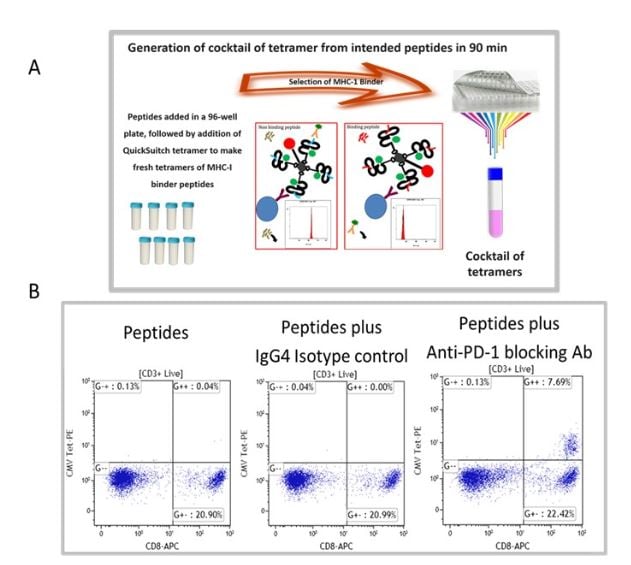
Figure 1. Improved recall antigen assay for function-based screening of immune checkpoint drug candidates. A shows the peptide exchange MHC tetramer platform (MBL International, Woburn MA), which allows the generation of MHC tetramers with intended recall antigen peptide and making a validated cocktail of tetramers corresponding to multiple recall antigens. The tetramers are pooled after individually being made and after desalting step to remove unbound peptides. We have validated this tetramer pool system and the staining pattern was not significantly different from manufactured tetramers for all tested peptides. B. Shows the result of a selected and characterized “PD-1 blockade responder”.
Currently, we select and bank the PBMC of validated super responders for functional screening of ICBs.
The PBMC of our characterized hosts post-stimulation response with a conditioned media are divided into responders and non-responders to immune checkpoint blockades. After stimulation with peptide and PD-1 blocking antibodies, Tetramer positive cells in PBMC of responders to PD-1 blockade express higher levels of PD-1, TIM-1, LAG-3, TOX and they have reduced IFN-g expression post antigen stimulation, their monocytes express increased PD-L1 levels.
We have optimized / maximized each element of the assay, the in vitro stimulation as well as the readout of the assay. For example, using an MHC tetramer guided Recall Antigen assay in characterized donors, we use CMV, EBV, and influenza selected peptide pool for stimulation, and as a readout we used a cocktail of tetramers and activation markers plus IFN-g secretion. PBMC samples characterized and validated as responders to antigen recall are then used for functional screening of ICB drug candidates. We use validated active ICB or co-stimulatory biologics as well as isotype antibodies as positive and negative controls, respectively. As shown in Figure 2, the improved recall antigen assay creates a window of TEX-like and / or suboptimal conditions that can be corrected by an active co-inhibitory blockade biologic (e.g. pembrolizumab).
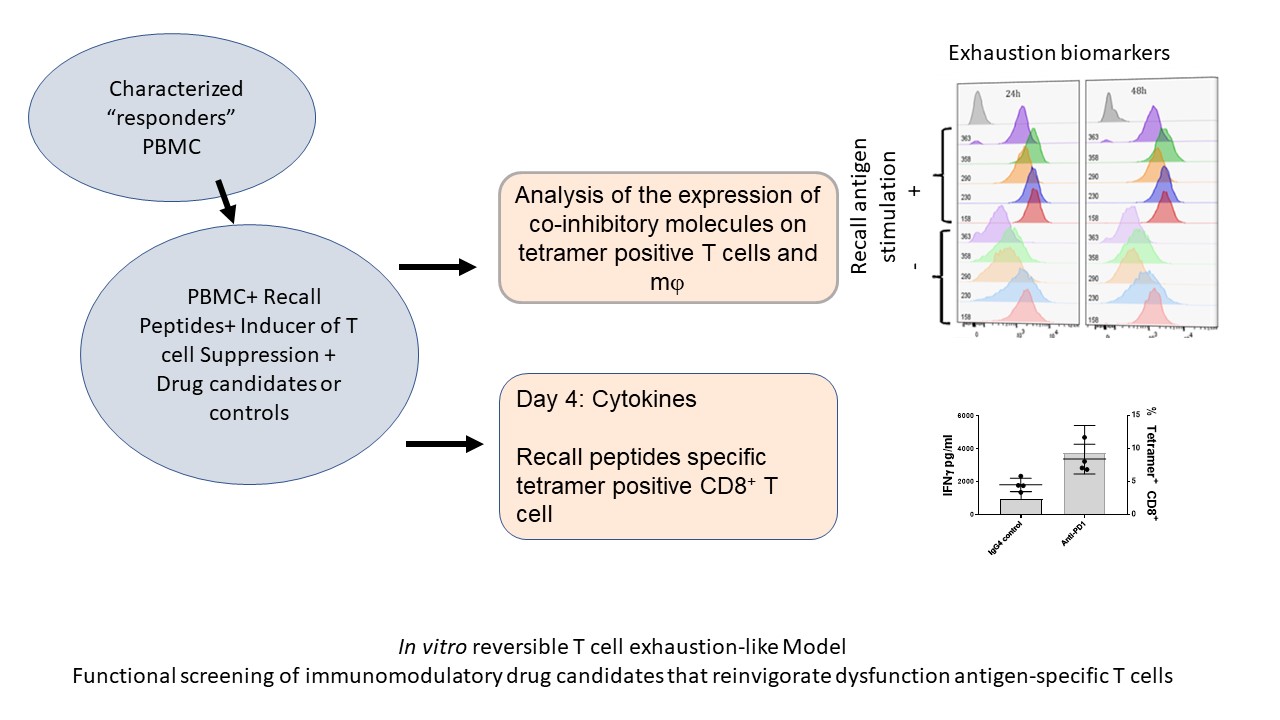
Figure 2. Depicts the process of reversible T cell dysfunction assay.
If you are interested in using this assay to screen your drug candidates, please click the button below to contact us!

References
- McLane, L.M., M.S. Abdel-Hakeem, and E.J. Wherry, CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol, 2019. 37: p. 457-495.
- Hashimoto, M., et al., CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med, 2018. 69: p. 301-318.
- Shin, H., et al., Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J Exp Med, 2007. 204(4): p. 941-9.
- Schietinger, A., et al., Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity, 2016. 45(2): p. 389-401.
- Crawford, A., et al., Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity, 2014. 40(2): p. 289-302.
- Yi, J.S., M.A. Cox, and A.J. Zajac, T-cell exhaustion: characteristics, causes and conversion. Immunology, 2010. 129(4): p. 474-81.
- Thommen, D.S. and T.N. Schumacher, T Cell Dysfunction in Cancer. Cancer Cell, 2018. 33(4): p. 547-562.
- Chang, C.H., et al., Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell, 2015. 162(6): p. 1229-41.
- Eil, R., et al., Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature, 2016. 537(7621): p. 539-543.
- Vodnala, S.K., et al., T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science, 2019. 363(6434).
- Ohta, A., A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front Immunol, 2016. 7: p. 109.
- Anderson, K.G., I.M. Stromnes, and P.D. Greenberg, Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell, 2017. 31(3): p. 311-325.
- Cullen, J.G., et al., CD4(+) T help promotes influenza virus-specific CD8(+) T cell memory by limiting metabolic dysfunction. Proc Natl Acad Sci U S A, 2019.
- Revenfeld, A.L.S., et al., Induction of a Regulatory Phenotype in CD3+ CD4+ HLA-DR+ T Cells after Allogeneic Mixed Lymphocyte Culture; Indications of Both Contact-Dependent and -Independent Activation. Int J Mol Sci, 2017. 18(7).
- Ghosh, S., et al., TSR-033, a Novel Therapeutic Antibody Targeting LAG-3, Enhances T-Cell Function and the Activity of PD-1 Blockade In Vitro and In Vivo. Mol Cancer Ther, 2019. 18(3): p. 632-641.
- Askar, M., et al., MHC Class I Chain-Related Gene A (MICA) Donor-Recipient Mismatches and MICA-129 Polymorphism in Unrelated Donor Hematopoietic Cell Transplantations Has No Impact on Outcomes in Acute Lymphoblastic Leukemia, Acute Myeloid Leukemia, or Myelodysplastic Syndrome: A Center for International Blood and Marrow Transplant Research Study. Biol Blood Marrow Transplant, 2017. 23(3): p. 436-444.
- Wang, C., et al., In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res, 2014. 2(9): p. 846-56.
- Grenga, I., et al., A fully human IgG1 anti-PD-L1 MAb in an in vitro assay enhances antigen-specific T-cell responses. Clin Transl Immunology, 2016. 5(5): p. e83.
- Li, Y., et al., Discovery and preclinical characterization of the antagonist anti-PD-L1 monoclonal antibody LY3300054. J Immunother Cancer, 2018. 6(1): p. 31.
- Agency, E.M., Guideline on potency testing of cell based immunotherapy medicinal products for the treatment of cancer. 2016.