
Published by Yuri Poluektov on
In our previous blog post (How to screen SARS-CoV-2 peptides to facilitate T cell research) we have described how the immune system samples all of the pathogenic proteins by looking at the small fragments of each protein and making a determination on whether that fragment (also referred to as peptide) belongs in the body. Not all peptides are presented to the immune system equally and most of the amino acids that make up the proteins of the pathogen remain completely invisible to our immune system. It is only a small portion of the entire amino acid sequence that gets examined by the immune system. That is why it is crucial to know which peptides from the pathogen you are trying to study are actually presented to the immune system, and, more importantly, which of those peptides are able to stimulate an immune response.
Peptide presentation is still an ongoing topic of study, with few definitive answers readily available. But there are a few tools for researchers trying to determine a peptide sequence of a pathogen’s proteins that is examined by the immune system and that could stimulate the T cells. One such tool is the Immune Epitope Database (IEDB). This web resource, funded by NIAID, catalogues experimental data and analyzes it to allow the prediction of peptide binding affinities for specific MHC molecules. As we accumulate more experimental data, this resource will become more and more effective at predicting the right peptide sequences.
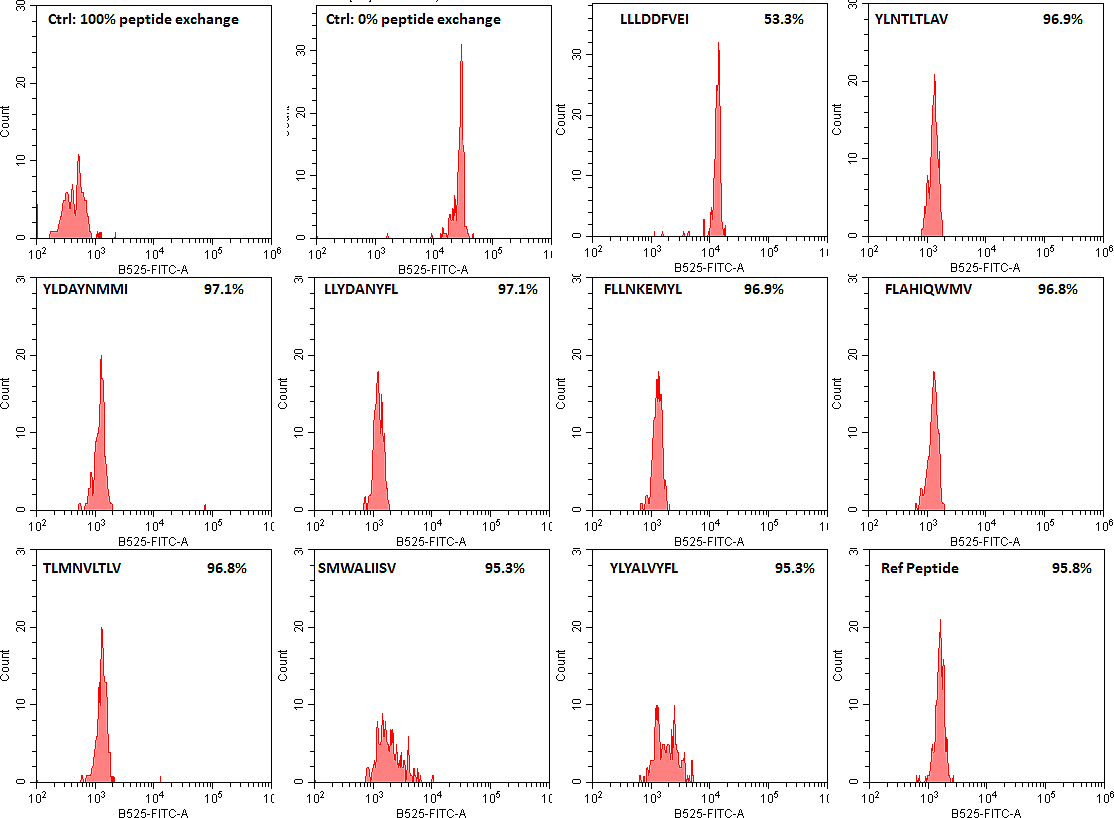
As a validation of our QuickSwitchTM MHC tetramer staining platform, we utilized the innate ability of our platform to determine the relative peptide binding affinity of SARS-CoV-2 derived peptides to the HLA-A*02:01 Class I MHC molecule and compared it to the theoretical binding affinity predicted by IEDB. While most of the peptides we have tested correlate well with their predicted binding affinities, there are a few outliers. While we examined only a small set of possible peptides, if we were to examine more peptides there would be a lot more outliers that the theoretical prediction algorithm would miss out on but that would be effectively presented by the immune system and vice versa. Someday the in silico prediction algorithms will become more robust, but, for now, all of the predicted results have to be verified by in vivo and in vitro experiments.
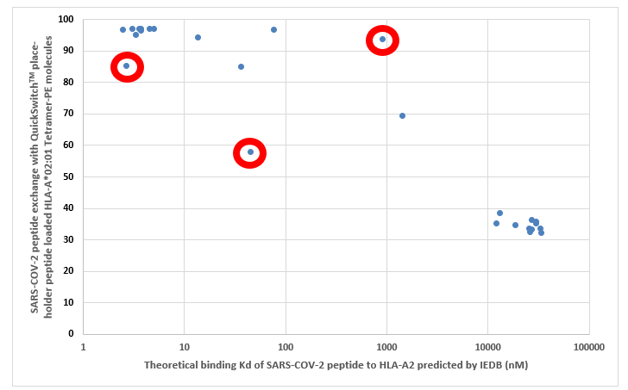
Theoretical binding affinity of SARS-CoV-2 peptides to HLA-A*02:01 compared to the practical binding to a Tetramerized recombinant MHC molecule. The peptides outlined in red circles are YLYALVYFL (Theoretical Kd = 2.67 nM, Practical Exchange rate = 85%); SVLLFLAFV (Theoretical Kd = 44.91 nM, Practical Exchange rate = 57.5 %) and ISDEFSSNV (Theoretical Kd = 908.9 nM, Practical Exchange rate = 93.5%)
Click the link below to view a full description of experiment from the Poluektov, Daftarian and Delcommenne manuscript.
